I have a github page which tracks some of the projects that I collaborate on. Probably it is more up to date than what you see here, but it does not correspond 1 to 1 in terms of the projects tracked.
If you use my code (or try and fail to use my code) then please send an email to the address on my uni.lu page saying roughly what you used it for and how you got on. Its always interesting for me to hear from people solving actual problems with my work.
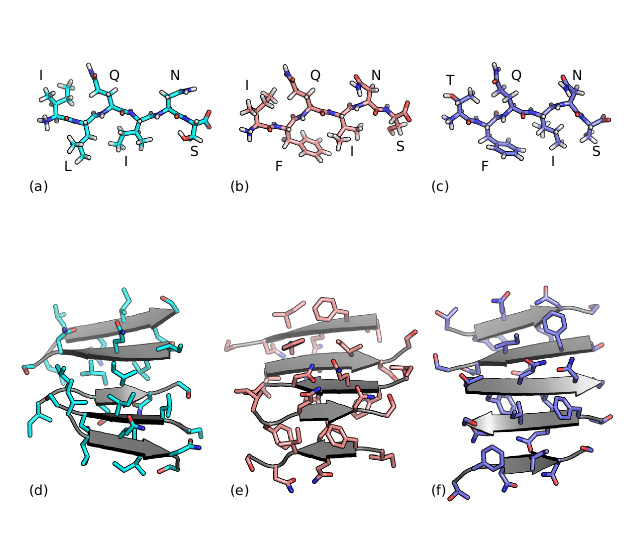
Running with a system of a few dozen peptides, Ali's code accelerates aggregation, generating multiple (thermally disordered) quasi-equilibrium aggregated all-atom structures. Most of the structures found were anti-parallel beta sheet, for the systems considered (see picture, left, taken from Ali's thesis).
The FRESHS (Flexible Rare Event Sampling Harness System) code is online here. This is a parallel execution toolkit to combine the SPRES and FFS sampling algorithms with the LAMMPS, Gromacs and ESPResSo simulation programs (or with your own self-written code).
Full text of the paper which gives an in-depth look at the SPRES algorithm is behind a paywall at J Chem Phys, however you will find an earlier version of the paper on arXiv.
To cite the FRESHS software package refer to this paper: "The Flexible Rare Event Sampling Harness System (FRESHS)".
I have written a library implementing the techniques documented in this open access J Chem Theory Comput article as a plugin for AMBER. This is now available as part of the AmberTools14 release.
I recently embarked on a rewrite of the tumbling cross-section code which has garnered about 250 citations since I knocked it together as a PhD student with the help of sundry individuals from Alison Ashcroft's group. The idea is very simple, just sample over orientations of the molecule to estimate tumbling cross section, thence to find the friction with a buffer gas as it moves through the spectrometer. Sometimes simple things are also interesting and fun.
This is just a dump of files that I want to be available for whatever lab assignment or workshop. If you are just browsing then you probably won't have a use for any of this stuff:
01_neural_20mins.zip 1EBY.zip 1EC0.zip 1W5V.zip 1W5W.zip 1W5X.zip 1W5Y.zip intro.pdf instructions.pdf